 CAREER: Exploring the transcriptional regulatory networks controlling the early stages of legume nodulation
CAREER: Exploring the transcriptional regulatory networks controlling the early stages of legume nodulation
Nodulation is a process that underlies the fixation of atmospheric nitrogen via symbioses of soil-borne microorganisms (i.e. rhizobia) with leguminous plant. This is an economically important process as it reduces the amount of nitrogen fertilizer needed to grow crops and is a key component in sustainable agriculture. Thus, a better understanding of the molecular mechanisms controlling the nodulation process should lead to ways to more effectively capture and fix atmospheric nitrogen and to the transfer of this capacity to non-legume plants. One notable achievement in our understanding of legume nodulation is the characterization of the signaling cascade that controls the infection of the plant root hair cell by rhizobia, the initial step of nodulation. This symbiotic pathway ends with several transcription factors such as Nodulation Signaling Pathway1 (NSP1) and the transcriptional regulation of almost 2000 genes in legume root hair cells upon rhizobial inoculation. The investigators proposes to characterize the NSP1 regulatory network, a network essential in controlling the early stages of nodulation. In addition to have a significant impact on legume research, this project also includes the development of unique educational programs dedicated to Oklahoma high-school and undergraduate students from under-represented groups, promoting strong integration between research and education.
This CAREER project is built on the hypothesis that regulatory networks directly under the control of one or several TFs of the symbiotic pathway orchestrate the proper activation and timing of gene expression in root hair cell in response to rhizobia. Specifically focusing on the Nodulation Signaling Pathway1 (NSP1) regulatory network, this project will specifically address two key questions: What is the identity of the soybean and medicago root hair genes directly under the control of NSP1? What are the protein complexes centralized around NSP1 and controlling the expression of root hair genes in response to rhizobial inoculation? To answer these questions, the investigators propose to 1) characterize the soybean root hair genes directly under the control of GmNSP1a and b in response to B. japonicum by taking advantage of high-throughput sequencing technologies and by applying DamID-seq or ChIP-seq methods; 2) identify the protein partners of GmNSP1a and b in root hair cells inoculated with B. japonicum after co-immunoprecipitation of the NSP1 protein partners in root hair cells upon rhizobia inoculation; 3) validate the conservation of the root hair gene regulatory networks between soybean and Medicago truncatula, the model legume.
 Use of a single cell type model, the root hair cell, to advance our understanding of the soybean and sorghum transcriptomic and epigenomic responses to various environmental stresses
Use of a single cell type model, the root hair cell, to advance our understanding of the soybean and sorghum transcriptomic and epigenomic responses to various environmental stresses
The dynamic changes of the epigenome in response to a stress and its impact on gene expression remain unclear. To clarify this relationship, we propose to characterize the epigenomic and transcriptomic responses of one single soybean and sorghum cell type, the root hair cell to various environmental stresses. The use of the same single cell type from two JGI flagship plants will enhance our understanding of the influence of various environmental stresses by characterizing both the variable and constant transcriptomes and epigenomes (i.e. DNA methylome and small RNAs) within and between plant species. The research proposed is novel since it focuses on a system biology approach at the level of one single plant cell type. Our choice to focus on one single differentiated plant cell type is motivated by the fact that -omics studies performed on an entire organ cannot provide an accurate picture of the molecular response occurring in response to a stress due to the multicellular complexity of the organ. In fact, -omic datasets generated from entire plant organs masks cell-specific characteristics of the transcriptome and epigenome and lead to a dilution of the transcripts and epigenomic changes [1]. In addition, the intimate interaction between the root hair cell and the rhizosphere makes it the first cell type to perceive changes in the rhizosphere environment. To be successful in this project, we recently developed an ultrasound aeroponic system allowing the formation of an abundance root hair cell population, their even treatment and the isolation of gram quantities of highly purified plant root hair cell.
 Comparative-, Functional-, and Epi-Genomics of Legumes and Nodule Formation
Comparative-, Functional-, and Epi-Genomics of Legumes and Nodule Formation
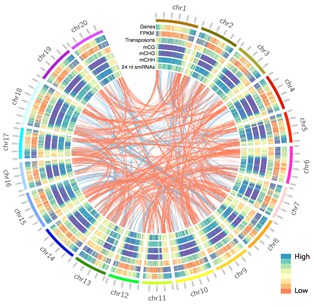
Legumes play an important and unique role in agriculture by undergoing nodulation, a process that through symbioses with soil-borne bacterial microorganisms enable legumes to capture and fix atmospheric nitrogen, providing a source of nitrogen to the plant and enhancing soil fertility. Nodulation begins with an exchange of bacterial and plant signals that results in the bacteria infecting the root hair cell, a root epidermal cell characterized by its polar growth, and leading to reprogramming of cellular and tissue development to form a specialized organ, the nodule. Despite the importance of nodulation, questions remain as to how signals perceived by the plant are translated into re-patterning of tissue development. Reprogramming the epigenome is essential for gene regulation, for plant/tissue/cell development and for successful bacterial infection of the plant cells. However, it is not yet known how plant epigenomes are reprogrammed during bacterial perception and nodule development in legumes.
By combining comparative and population epigenomic approaches, this proposal aims to: i) identify population-wide sets of natural epigenetic variants regulated by RNA-directed DNA methylation (RdDM); and ii) determine how plant epigenomes are reprogrammed during the initial perception of bacteria and during development of nodules in samples enriched for single cell types. RdDM loci are thought to be sensitive to external stimuli (e.g. stress and bacteria) and this will be done within and among Medicago truncatula, Phaseolus vulgaris and Glycine max and will provide a resource of novel natural variants to these research communities. For nodulation, methods will be used to enrich for single cell types, such as root hair cells and infected cells of the nodule, to overcome challenges of multi-cellular complexity, which plague attempts to investigate relationships among gene expression, transcription factor (TFs) binding, and epigenomes. Consequently, this proposal will lead to a more complete and fundamental understanding of legume epigenomes and epigenomic reprogramming that occurs throughout the nodulation process and specifically addresses the 2013 focus area: “Systems-level approaches to understand the interaction between the genome and the epigenome in the regulation of economically important processes in crop plants.
A more fundamental understanding of nodulation will lead to the ability to better engineer this process to more effectively capture and fix atmospheric nitrogen. In addition to this agricultural impact, a continuation of a long-term partnership with primarily Hispanic and Native American tribal colleges in New Mexico will occur, where partnerships are formed with these institutions to provide training through workshops and summer internships for students and faculty interested in research careers. Three graduate students, one in each lab, will be trained and we include yearly workshops for these students that will focus on specific aspects of plant biology as it relates to this project. These workshops will be open to all graduate students at the respective institutions.
 Genomics of Energy Sorghum's Water Use Efficiency/Drought Resilience
Genomics of Energy Sorghum's Water Use Efficiency/Drought Resilience
The proposed research will utilize genetic variation in sorghum germplasm to identify traits-QTL-gene regulatory networks that enhance water use efficiency, soil water extraction, and drought resilience. Potentially useful leaf and root traits have been identified from prior research, analysis of energy sorghum under water limited field conditions, and through crop-trait modeling. Water use efficiency, soil water extraction by roots, drought resilience and associated canopy/root traits will be analyzed using lysimeters, aeroponics, and irrigated/non irrigated field plots. An energy sorghum association panel and three RIL populations will be screened for variation in target traits. Quantitative genetics and genomic approaches will be used to identify QTL and alleles that modify trait expression and to characterize target trait-gene regulatory networks. Near isogenic lines (NILs) varying in target trait alleles will be constructed during HIF-based QTL fine mapping and allele identification for molecular studies and field analysis. Particularly important trait-gene regulatory networks will be subject to more detailed molecular characterization using RNAseq-aided transcriptome analysis of NILs. Information from molecular genetic analysis will enable targeted mining of germplasm resources for additional genetic variation, the development of improved hybrids, and provide information needed for engineering enhanced genetic designs through transgenics or directed mutagenesis.
 Unraveling the Transcriptional Regulation of Plant Cell Elongation
Unraveling the Transcriptional Regulation of Plant Cell Elongation
The objective of this proposal is to characterize the transcriptional regulatory mechanisms controlling plant cell elongation. This project is built on the following hypothesis: transcription factors (TFs) act as hubs regulating the expression of hundreds of genes to control plant cell elongation. During the two years of funding, we propose to accomplish the following objectives:
Characterization of the transcriptomic profile of one single plant cell type, the soybean root hair cell, at various stages of its elongation process. Taking advantage of the release of the soybean genome sequences and development of Illumina high-throughput sequencing (HTS) technology, we will characterize the transcriptome of synchronized soybean root hair cells at different stages of the elongation process. The soybean root hair cell which is characterized by its polar elongation is selected as the model for this project based on the development of methods to control and synchronize root hair cell elongation maximizing the homogeneity of the plant material and the ease of access to gram quantities of this synchronized single cell type compatible with âomics analyses including transcriptional analysis.
Also, working with one single cell type model will provide a deeper and more systematic understanding of plant cell elongation processes. We foresee that this transcriptomic analysis will enable the identification of hundreds of genes differentially regulated during the stages of plant cell elongation, including several TF genes.